8 SNPTEST
A simple association test use case will be illustrated in this section to portrait the usage of SNPTEST in OmicSHIELD.
|
⚠️ RESOURCES USED ALONG THIS SECTION |
||||||
|---|---|---|---|---|---|---|
|
From https://opal-demo.obiba.org/ : |
||||||
|
The structure followed is illustrated on the following figure.
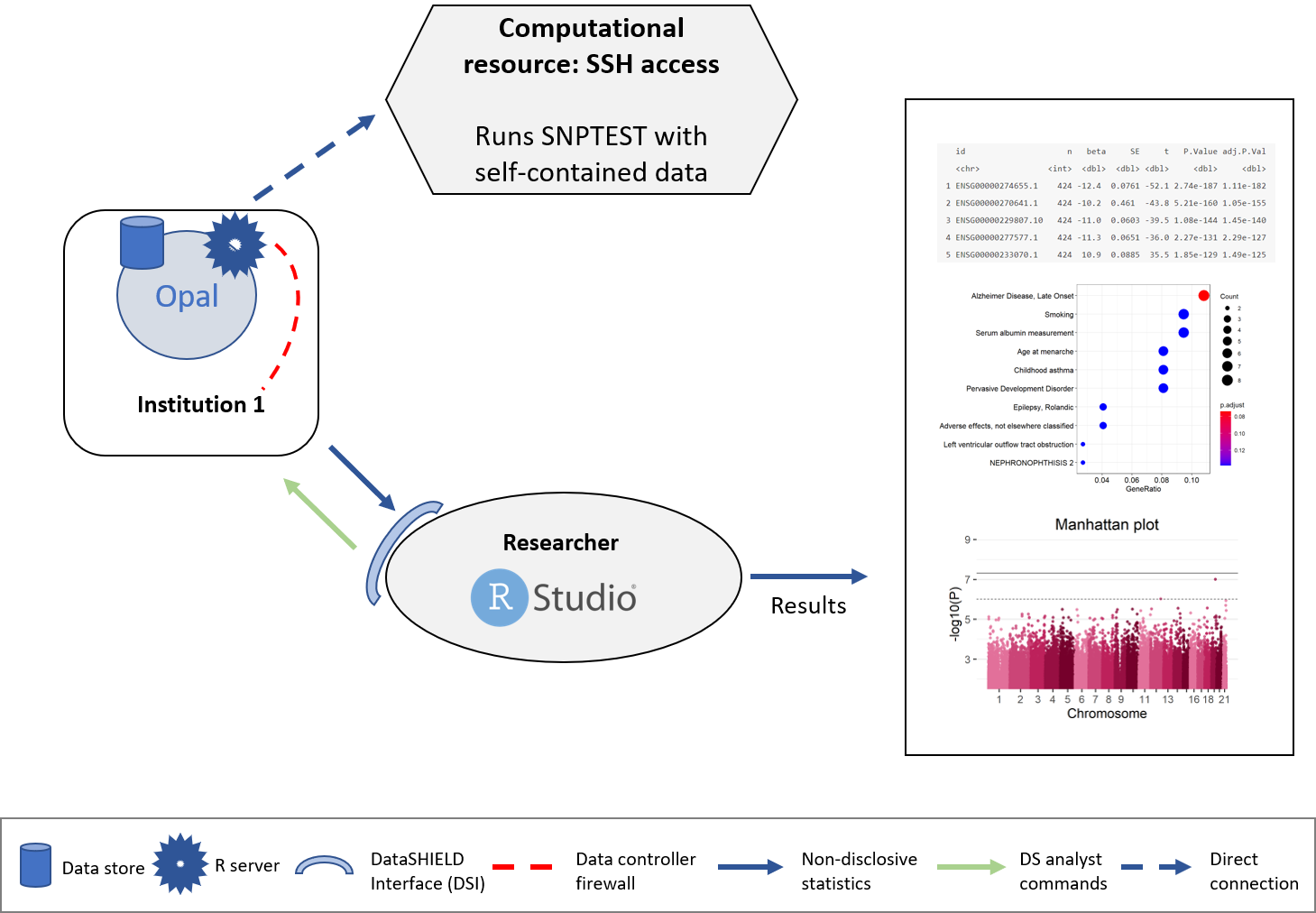
Figure 8.1: Proposed infrastructure for SNPTEST analysis.
The data analyst corresponds to the “RStudio” session, which through DataSHIELD Interface (DSI) connects with the Opal server located at the cohort network. This Opal server contains a resource with the URL where the computational resource is located, also the access credentials are kept secret inside the resource and only the Opal administrator has access to them. A connection to this computational resource is created on the Opal server and the aggregated results are passed to the analyst.
8.1 Connection to the Opal server
We have to create an Opal connection object to the cohort server. We do that using the following functions.
library(DSOpal)
library(dsBaseClient)
library(dsOmicsClient)
builder <- newDSLoginBuilder()
builder$append(server = "study1", url = "https://opal-demo.obiba.org",
user = "dsuser", password = "P@ssw0rd",
resource = "RSRC.brge_snptest",
profile = "omics")
logindata <- builder$build()
conns <- datashield.login(logins = logindata, assign = TRUE,
symbol = "client")8.2 Build the SNPTEST call
Now, we are ready to run any SNPTEST command from the client site. Notice that in this case we want to assess association between the genotype data in gen format and use as phenotype the variable ‘bin1’ that is in the file ‘sample’, which is a binomial variable (case/control). The sentence in a SNPTEST command would be (NOTE: we avoid -o to indicate the output file since the file will be available in R as a tibble). The arguments must be encapsulated in a single character without the command ‘snptest’.
snptest.arguments <- "-frequentist 1 -method score -pheno bin1 -data cohort1.gen cohort1.sample cohort2.gen cohort2.sample"8.3 Call the analysis
Then, the analyses are performed by:
ans.snptest <- ds.snptest("client", snptest.arguments)The object ans.snptest contains the SNPTEST results at each server as well as the outuput provided by the command
lapply(ans.snptest, names)$study1
[1] "results" "snptest.out"head(ans.snptest$study1$results)# A tibble: 6 x 50
alternate_ids rsid chromosome position alleleA alleleB index average_maximum~
<chr> <chr> <lgl> <dbl> <chr> <chr> <dbl> <dbl>
1 INSERTION_1 RSID~ NA 1 A AGTGCTA 1 0.992
2 DELETION_1 RSID~ NA 2 A - 2 0.946
3 SNPID_3 RSID~ NA 3 A G 3 0.986
4 SNPID_4 RSID~ NA 4 A G 4 0.991
5 SNPID_5 RSID~ NA 5 A G 5 0.984
6 SNPID_6 RSID~ NA 6 A G 6 0.992
# ... with 42 more variables: info <dbl>, cohort_1_AA <dbl>, cohort_1_AB <dbl>,
# cohort_1_BB <dbl>, cohort_1_NULL <dbl>, cohort_2_AA <dbl>,
# cohort_2_AB <dbl>, cohort_2_BB <dbl>, cohort_2_NULL <dbl>, all_AA <dbl>,
# all_AB <dbl>, all_BB <dbl>, all_NULL <dbl>, all_total <dbl>,
# cases_AA <dbl>, cases_AB <dbl>, cases_BB <dbl>, cases_NULL <dbl>,
# cases_total <dbl>, controls_AA <dbl>, controls_AB <dbl>, controls_BB <dbl>,
# controls_NULL <dbl>, controls_total <dbl>, all_maf <dbl>, cases_maf <dbl>,
# controls_maf <dbl>, missing_data_proportion <dbl>, het_OR <lgl>,
# het_OR_lower <lgl>, het_OR_upper <lgl>, hom_OR <lgl>, hom_OR_lower <lgl>,
# hom_OR_upper <lgl>, all_OR <lgl>, all_OR_lower <lgl>, all_OR_upper <lgl>,
# frequentist_add_pvalue <dbl>, frequentist_add_info <dbl>,
# frequentist_add_beta_1 <dbl>, frequentist_add_se_1 <dbl>, comment <chr>ans.snptest$study$snptest.out$status
[1] 0
$output
[1] "Welcome to SNPTEST v2.5.2 (revision 2a8e744975fbd46cc3c020721719dd415da0cb89)"
[2] "© University of Oxford 2008-2015"
[3] "https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html"
[4] "Read LICENCE file for conditions of use."
[5] ""
[6] "=============="
[7] ""
[8] "Data Files : "
[9] " -gen files : cohort1.gen cohort2.gen "
[10] " -sample files : cohort1.sample cohort2.sample "
[11] ""
[12] "Tests : "
[13] " -frequentist : 1"
[14] " -method score"
[15] ""
[16] "reading sample exclusion lists"
[17] ""
[18] "Inspecting data (this may take some time)..."
[19] "Sample and exclusions summary :"
[20] " - Number of individuals in : (cohort 1) (cohort 2) "
[21] " 500 500 "
[22] ""
[23] ""
[24] "Reading sample files :"
[25] "Summary of covariates and phenotypes"
[26] " # discrete variables : 3"
[27] " cov1 : type = D (Discrete covariate)"
[28] " cov2 : type = D (Discrete covariate)"
[29] " sex : type = D (Discrete covariate)"
[30] " # continuous variables : 2"
[31] " cov3 : type = C (Continuous covariate)"
[32] " cov4 : type = C (Continuous covariate)"
[33] " # phenotypes : 4"
[34] " pheno1 : type = P (Continuous phenotype)"
[35] " pheno2 : type = P (Continuous phenotype)"
[36] " bin1 : type = B (Binary phenotype)"
[37] " bin2 : type = B (Binary phenotype)"
[38] "Covariate summary :"
[39] " (no covariates in use.)"
[40] "Phenotype summary :"
[41] " bin1 : missing levels"
[42] " 2 1(998)"
[43] ""
[44] "You have specified the following model:"
[45] " bin1 ~ (baseline) + (genotype)"
[46] ""
[47] "Phenotype being used : bin1"
[48] ""
[49] "Data Summaries : "
[50] " -number of SNPs = (unknown)"
[51] ""
[52] "Data with missing genotype data threshold and exclusion list applied :"
[53] " cohort1.gen : 500"
[54] " cohort2.gen : 500"
[55] ""
[56] "--------------------------------------------------------------------------"
[57] ""
[58] "SinglePhenotypeTest"
[59] "--------------------------------------------------------------------------"
[60] ""
[61] "Analyzing Data :"
[62] " scanning... read chunk [1 of (unknown)]... done."
[63] " scanning... read chunk [2 of (unknown)]... done."
[64] " scanning... no more data."
[65] ""
[66] "finito"
$error
character(0)
$command
[1] "cd /home/master/data && snptest -frequentist 1 -method score -pheno bin1 -data cohort1.gen cohort1.sample cohort2.gen cohort2.sample -o /tmp/ssh-8008/ex.out"
attr(,"class")
[1] "resource.exec"datashield.logout(conns)