8 Extension to VCF files to peform GWAS with Bioconductor
Genomic data can be stored in different formats. PLINK and VCF files are commonly used in genetic epidemiology studies. In order to deal with this type of data, we have extended the resources available at the resourcer package to VCF files. NOTE: PLINK files can be translated into VCF files using different pipelines. In R you can use SeqArray to get VCF files.
We use the Genomic Data Storage (GDS) format which efficiently manages VCF files in the R environment. This extension requires creation of a Client and a Resolver function for the resourcer that are located in the dsOmics package. The client function uses snpgdsVCF2GDS function implemented in SNPrelate to coerce the VCF file to a GDS object. Then the GDS object is loaded into R as an object of class GdsGenotypeReader from GWASTools package that facilitate downstream analyses.
The Opal server API allows us to incorporate this new type of resource as illustrated in Figure 8.1.
Figure 8.1: Description of how a VCF file can be added to the opal resources
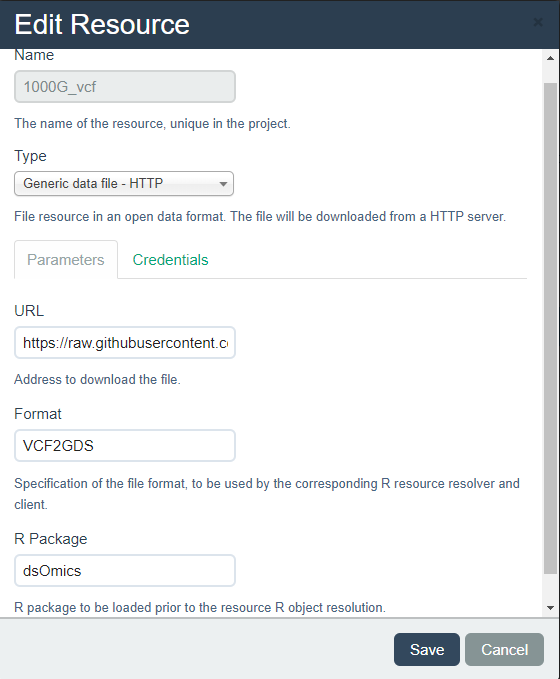
It is important to notice that the URL should contain the tag method=biallelic.only&snpfirstdim=TRUE since these are required parameters of the snpgdsVCF2GDS function. This is an example:
https://raw.githubusercontent.com/isglobal-brge/scoreInvHap/master/inst/extdata/example.vcf?method=biallelic.only&snpfirstdim=TRUEIn this case we indicate that only biallelic SNPs are considered (‘method=biallelic.only’) and that genotypes are stored in the individual-major mode, (i.e., list all SNPs for the first individual, and then list all SNPs for the second individual, etc) (‘snpfirstdim=TRUE’).