2 Pre-processing of DNA methylation data
In this chapter will explain the steps for the pre-processing of
DNA methylation data with the meffil R package. A file with the slides
can be downloaded here
At the end of the practice, please answer these questions:
- Is there any sample that is excluded due to the meth vs unmeth comparison (low-quality samples)?
- Is there any sample that is excluded due to inconsistent sex?
- Which are the main biological and technical variables associated with PC1?
2.1 Creation of the Sample Sheet
In these first steps we will work with a little subset of the data just to show how the code works and be able to run the commands in short time. Afterwards, we will load the complete data to keep going with the analyses.
These are the libraries that will be used:
Then we need to create a sample sheet from the IDAT files (in this case, 20 IDAT files corresponding to 10 individuals)
samplesheet_sub <- meffil.create.samplesheet("./data/DAY2/Input/IDAT_cohort1_subset",
recursive=TRUE)
dim(samplesheet_sub)## [1] 10 6The function automatically adds the sample name, slide, row, column and filename from the IDATs. We can not remove these columns but we can manually add more. In this case we add the sex, age and smoking status variables. If we had duplicated samples for the same individual, we would also add the individual ID and then remove the duplicates.
First we load the phenotype data
pheno <- read.table("./data/DAY2/Input/GSE42861_pheno_cohort1.txt",
sep = '\t', header = T)
head(pheno)And then add the sex, the age and the smoking status variables, keeping only those samples with metadata
samplesheet_sub <- merge(samplesheet_sub[,c('Sample_Name', 'Slide',
'sentrix_row', 'sentrix_col', 'Basename')],
pheno[,c('Sample', 'Age', 'Sex', 'Smoking')],
by.x = 'Sample_Name',
by.y = 'Sample')
head(samplesheet_sub)2.2 Sample and CpG quality control
Once we have the sample sheet, the first step for the QC is the creation of the QC objects. We specify the cell type reference (idol optimized) and the thresholds for detection and minimum number of beads.
# DO NOT RUN
meffil.list.cell.type.references()
qc.objects <- meffil.qc(samplesheet_sub,
cell.type.reference="blood idoloptimized",
verbose=TRUE,
detection.threshold = 0.01,
bead.threshold = 3)As you can see, this step is very time-consuming.
Now that we have learned to create the SampleSheet and run the first steps of the QC using 10 samples, we will continue working with the full dataset. We need to load the same QC Objects we have just created, but those including all the samples
load('./data/DAY2/Output/QCObjects_cohort1.RData')After that, we setup the parameters for the QC
qc.parameters <- meffil.qc.parameters(
beadnum.samples.threshold = 0.05,
detectionp.samples.threshold = 0.03,
detectionp.cpgs.threshold = 0.05,
beadnum.cpgs.threshold = 0.05,
sex.outlier.sd = 3
)And run it
qc.summary <- meffil.qc.summary(
qc.objects,
parameters = qc.parameters,
genotypes=NULL,
verbose=TRUE
)DO NOT RUN THIS STEP IN THE VIRTUAL MACHINE - The report can be found here
This line creates an html report with several QC tests. We need to examine it with detail.
meffil.qc.report(qc.summary,
output.file="./data/DAY2/Output/QCReport_cohort1_student.html")If you are working in the virtual machine, just open the report that we have already created with this same data.
The next step is getting rid of the samples that do not pass the QC. In this case, after looking at the report, we have decided to maintain those samples that are outliers for the XY ratio.
outlier <- qc.summary$bad.samples
outlier <- outlier[outlier$issue!='X-Y ratio outlier',]
outlier
qc.objects <- meffil.remove.samples(qc.objects, outlier$sample.name)The probes that do not pass the QC will be removed afterwards during the normalisation step.
2.3 Normalisation
First of all we assess the number of needed PCs with the help of this plot:
y <- meffil.plot.pc.fit(qc.objects)
y$plot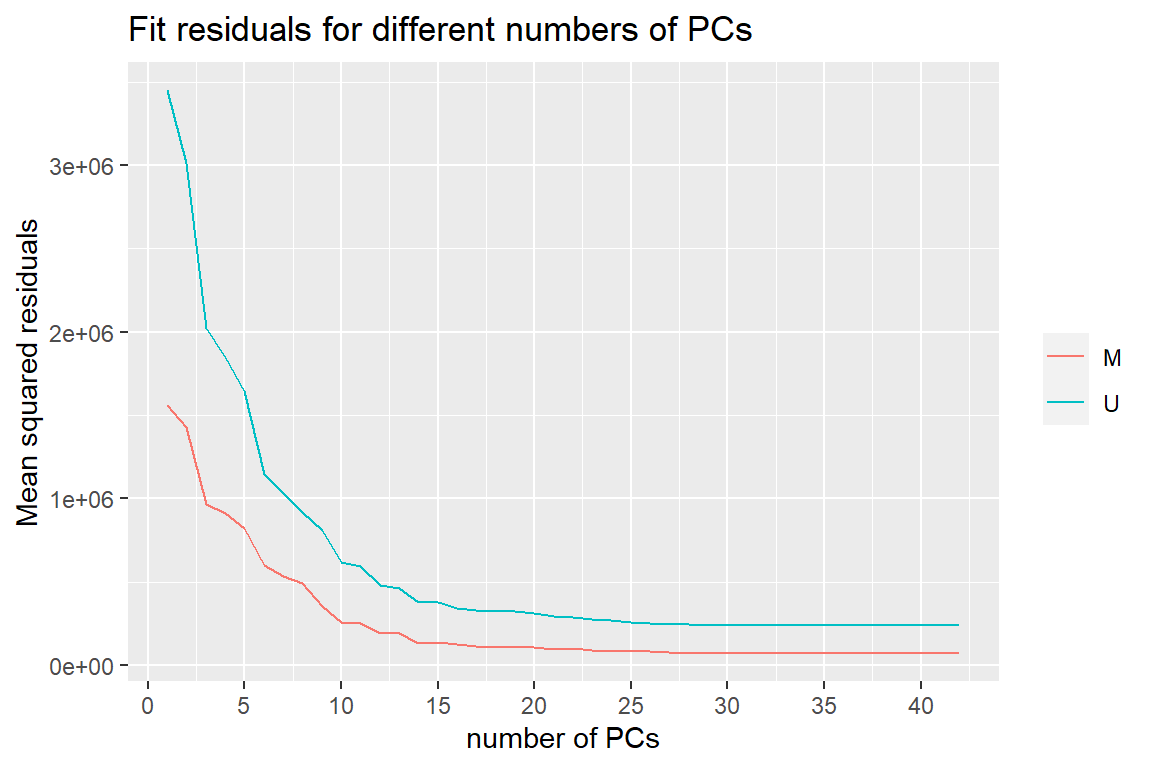
Then we perform the functional normalisation indicating the number of PCs and the IDs of bad CpGs from the QC to remove. After this step it is recommended to check again the missingness per sample and cpg
DO NOT RUN THOSE STEPS IN THE CLASS - they are very time consuming.
# DO NOT RUN
norm.objects <- meffil.normalize.quantiles(qc.objects, number.pcs=15)
# DO NOT RUN
norm.beta <- meffil.normalize.samples(
norm.objects,
just.beta=T,
remove.poor.signal = T,
cpglist.remove=qc.summary$bad.cpgs$name,
verbose=T)
# missing per sample - DO NOT RUN
miss_IDs <- miss_case_summary(data.frame(t(norm.beta)), order = TRUE)
miss_IDs$case <- colnames(norm.beta)
gc(reset=TRUE)
bad_ids <- miss_IDs[which(miss_IDs$pct_miss>5),]$case
# missing per cpg - DO NOT RUN
bad_cpgs <- miss_cpgs[which(miss_cpgs$pct_miss>5),]$variable
length(bad_cpgs)
# remove - DO NOT RUN
norm.beta <- norm.beta[!rownames(norm.beta) %in% bad_cpgs, !colnames(norm.beta) %in% bad_ids]
norm.objects <- norm.objects[!names(norm.objects)%in% bad_ids]
gc(reset=TRUE)Instead, load the normalisation objects and the betas that we provide:
load("./data/DAY2/Output/NormObjects_cohort1.RData")
load("./data/DAY2/Output/NormBeta_cohort1.Robj")Now we can create a normalisation report. We need to convert to factor all the batch variables that we want to assess and recalculate the PCs
for (i in 1:length(norm.objects)){
norm.objects[[i]]$samplesheet$Slide<-as.factor(norm.objects[[i]]$samplesheet$Slide)
norm.objects[[i]]$samplesheet$Sex<-as.factor(norm.objects[[i]]$samplesheet$Sex)
norm.objects[[i]]$samplesheet$Age<-as.factor(norm.objects[[i]]$samplesheet$Age)
norm.objects[[i]]$samplesheet$Smoking<-as.factor(norm.objects[[i]]$samplesheet$Smoking)
}
batch_var<-c("Slide","Sex", "Age", "Smoking")
norm.parameters <- meffil.normalization.parameters(
norm.objects,
variables=batch_var,
control.pcs=1:5,
batch.pcs=1:5,
batch.threshold=0.01
)
pcs <- meffil.methylation.pcs(norm.beta,verbose=T, full.obj = T)Finally, we obtain the normalisation summary and report.
Same as above, DO NOT RUN THIS STEP IN THE VIRTUAL MACHINE. The report can be found here
norm.summary <- meffil.normalization.summary(norm.objects,
pcs$x,
parameters=norm.parameters)
meffil.normalization.report(norm.summary,
output.file="./data/DAY2/Outout/NormReport_cohort1_student")2.4 Creation of a Expression Set
2.4.1 Estimation of cell type proportions
To run the EWAS, we will need to know the cell type proportions. They are automatically calculated in the first steps of the QC and can be recovered this way:
cc<-t(sapply(qc.objects, function(obj) obj$cell.counts$counts))
cc<-data.frame(PatientID=row.names(cc),cc) We add this information to the phenotype that we already have
phenoData <- merge(pheno, cc[,2:7], by.x = 'Sample', by.y = 0)
rownames(phenoData) <- phenoData[,1]
phenoData <- phenoData[,-1]
head(phenoData)2.4.2 Assembling
To assemble the final ExpressionSet we first create the metadata for the phenotypes, which consists on a dataframe with the variables description. Then we create an Annotated Dataframe.
mData <- data.frame(labelDescription=c("Sample cell type",
"Patient Age",
"Patient Sex",
"Smoking Status (never/current/former)",
"Percentage of B Lymphocytes",
"Percentage of CD4T Lymphocytes",
"Percentage of CDBT Lymphocytes",
"Percentage of Monocytes",
"Percentage of Neutrophils",
"Percentage of Natural Killer"),
row.names=colnames(phenoData))
phenoData = new("AnnotatedDataFrame", data=phenoData, varMetadata=mData)After that, we join the matrix of the normalized betas with the annotated dataframe of the phenotypes and cellular compositions. We have to make sure that the sample IDs coincide in the two datasets.
## [1] TRUE
eSet<-ExpressionSet(assayData=norm.beta,
phenoData=phenoData)
eSet## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 483153 features, 294 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: GSM1051533 GSM1051534 ... GSM1052213 (294 total)
## varLabels: Celltype Age ... NK (10 total)
## varMetadata: labelDescription
## featureData: none
## experimentData: use 'experimentData(object)'
## Annotation:Once we have saved the ExpressionSet with the normalised betas and the phenotypes we are ready to run the EWAS.
ANSWERS
- Yes. Samples GSM1051809, GSM1052037 and GSM1052049
- No. There are some sex outliers but there are no inconsistences
- Age (biological) and Slide (technical)