1 Resources and tools for epigenetics in Bioconductor
This section offers a summary of the main data structures that are implemented in Bioconductor for dealing epigenomic data data. Omic data is typically composed of three datasets: one containing the actual high-dimensional data of omic variables per individuals, annotation data that specifies the characteristics of the variables and phenotypic information that encodes the subject’s traits of interest, covariates and sampling characteristics. Bioconductor can effectively deal with this data using (among others) ExpressionSet objects which is a data structure that contains the beta values of individuals at each CpG, their genomic information and the phenotypes of the individuals. Specific data is accessed, processed and analyzed with specific functions from diverse packages, conceived as methods acting on the ExpressionSet object.
We will need these libraries:
library(BiocManager)
library(GenomicRanges)
library(GEOquery)
library(brgedata)1.1 Bioconductor
Bioconductor: Analysis and comprehension of high-throughput genomic data
- Statistical analysis: large data, technological artifacts, designed experiments; rigorous
- Comprehension: biological context, visualization, reproducibility
- High-throughput
- Sequencing: RNASeq, ChIPSeq, variants, copy number, …
- Microarrays: expression, SNP, …
- Flow cytometry, proteomics, images, …
Packages, vignettes, work flows
- 2230 packages (Apr’23); including …
- ‘Software’ packages – statistical methods, pipelines, …
- ‘Annotation’ packages – static data bases of identifier maps, gene models, pathways, etc; e.g., TxDb.Hsapiens.UCSC.hg19.knownGene
- ’Experiment packages – data sets used to illustrate software functionality, e.g., airway
- Discover and navigate via biocViews
- Package ‘landing page’
- Title, author / maintainer, short description, citation, installation instructions, …, download statistics
- All user-visible functions have help pages, most with runnable examples
- ‘Vignettes’ an important feature in Bioconductor – narrative documents illustrating how to use the package, with integrated code
- ‘Release’ (every six months) and ‘devel’ branches
- Support site; videos; recent courses.
- Common Workflows
Package installation and use
-
A package needs to be installed once, using the instructions on the package landing page (e.g., DESeq2).
require(BiocManager) install("DESeq2") # or BiocManager::install("DESeq2") -
older versions can be installed by
BiocManager::install("DESeq2", version = "3.8") -
Github packages can be install by
devtools::install_github("isglobal-brge/SNPassoc") -
Once installed, the package can be loaded into an R session
and the help system queried interactively, as outlined above:
1.2 ExpressionSet and GenomicRatioSet (and others: GRanges)
ExpressionSetwas one of the first implementations of Bioconductor to manage omic experiments. This figure illustrates how it is implemented
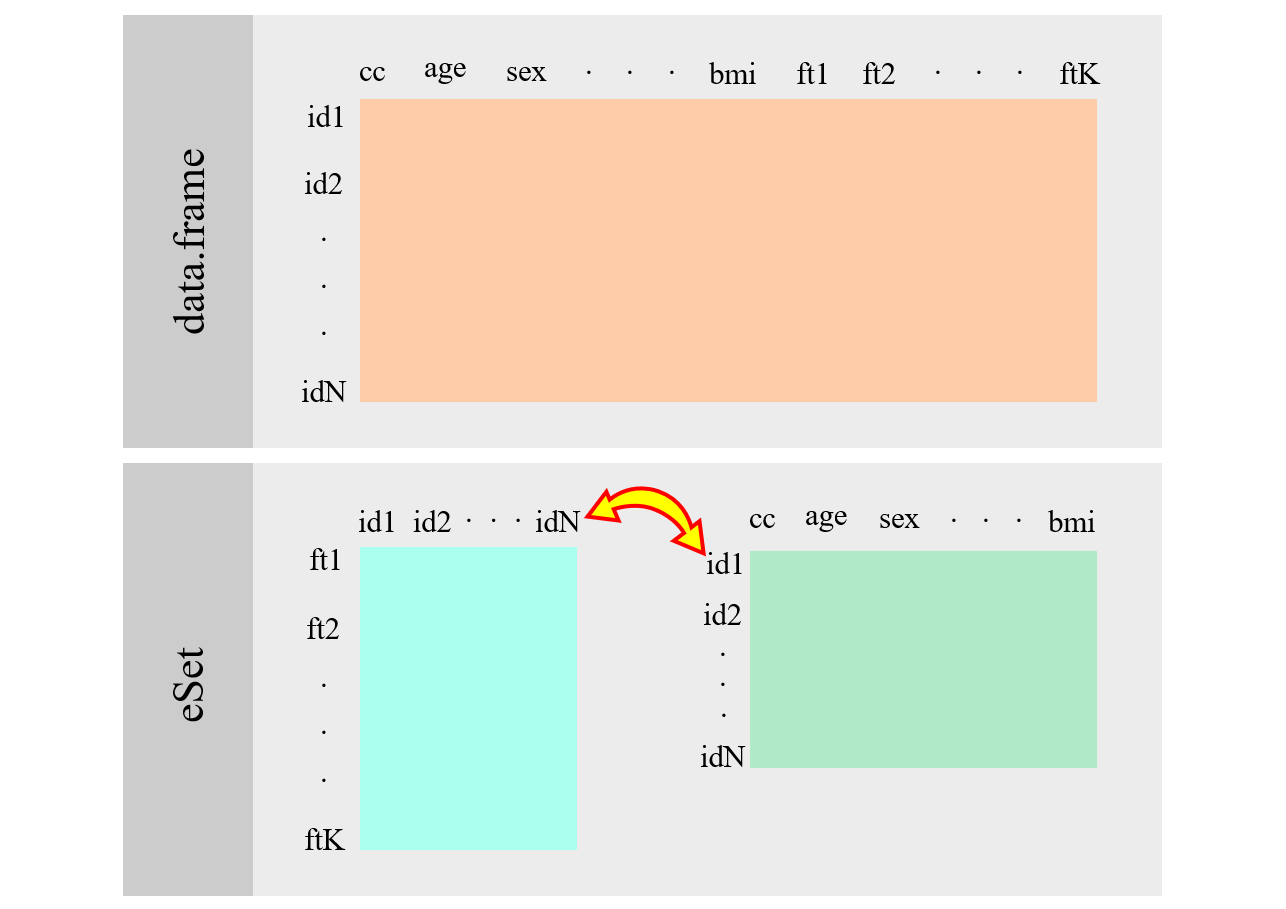
Figure 1.1: ExpressionSet scheme.
Although its use is discouraged in Bioconductor’s guidelines for the development of current and future packages (Summarized Experiments are preferred), most publicly data are available in this structure while future packages are still required to be able to upload and operate with it. This is the case of GEO (Gene Expression Omnibus) which is an international public repository that hosts and freely distribute an extensive collection of high-throughput gene expression data generated through technologies like microarrays and RNA sequencing.
1.2.1 Gene Expression Omnibus (GEO) repository
Researchers from diverse fields can upload their data to GEO, making it accessible to the scientific community for further analysis and interpretation. One of the notable benefits of GEO is its role in validating results from other studies. By comparing and contrasting their findings with existing datasets, researchers can corroborate their conclusions and ensure the reproducibility of their experiments. This validation process is essential for building a robust foundation of knowledge in molecular biology and fostering scientific progress through collaborative efforts and data-driven insights.
GEO was initially designed to deal with transcriptomic data, but it can also be used as a repository for epigenomic data. For example, we can further investigate the methylation differences in cases diagnoses with Alzheimer’s disease and controls using publicly available data. The study with accession number GSE80970, is a case/control (\(N=148/138\)) study of epigenomic data, measured with the Illumina Infinium Human 450K Methylation Array. Data is retrieved directly from the GEO website with the accession number.
Data for specific accessions in GEO can be downloaded in R using the getGEO of the Bioconductor’s package GEOquery
library(GEOquery)
if(!dir.exists("c:/tmp"))
dir.create("c:/tmp")
gsm <- getGEO("GSE80970", destdir = "c:/tmp")## Found 1 file(s)## GSE80970_series_matrix.txt.gz## Using locally cached version: c:/tmp/GSE80970_series_matrix.txt.gz## Using locally cached version of GPL13534 found here:
## c:/tmp/GPL13534.soft.gz
gsm <- gsm[[1]]This function downloads a data file called GSE80970\_series\_matrix.txt.gz that is retrieved automatically in an ExpressionSet object (this objects will be explain latter) that we have named gms.expr
class(gsm)## [1] "ExpressionSet"
## attr(,"package")
## [1] "Biobase"
gsm## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 485577 features, 286 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: GSM2139163 GSM2139164 ... GSM2139448 (286 total)
## varLabels: title geo_accession ... tissue:ch1 (45 total)
## varMetadata: labelDescription
## featureData
## featureNames: cg00000029 cg00000108 ... rs9839873 (485577 total)
## fvarLabels: ID Name ... SPOT_ID (37 total)
## fvarMetadata: Column Description labelDescription
## experimentData: use 'experimentData(object)'
## pubMedIds: 29550519
## Annotation: GPL13534An object of class ExpressionSet stores different tables including the expression profiles for each probe and subject assayData, phenotype data with traits measurements and covariates of interest pData, and feature data with information about the probe’s used in the expression (or methylation) array fData (e.g. annotation). Specific data is retrieved using the necessary functions. In particular, exprs () and phenoData () extract data tables for subjects’ expression levels and phenotypes/covariates, respectively. There are three other slots protocolData, experimentData and annotation (that uses Bioconductor databases as annotation data - i.e. it does not requires fData) that specify equipment-generated information about protocols, resulting publications and the platform on which the samples were assayed. Methods are implemented to extract the data from each slot of the object.
Let us see how it works. The function exprs () extracts the epigenetic data in a matrix where subjects are columns and probes are rows
## [1] 485577 286
expr[1:10, 1:5]## GSM2139163 GSM2139164 GSM2139165 GSM2139166 GSM2139167
## cg00000029 0.59167 0.61134 0.52240 0.55689 0.66928
## cg00000108 0.86955 0.87228 0.82742 0.89817 0.88524
## cg00000109 0.71494 0.68538 0.74456 0.76460 0.74441
## cg00000165 0.28111 0.26410 0.23158 0.27409 0.27160
## cg00000236 0.81495 0.78115 0.80187 0.80274 0.83936
## cg00000289 0.49322 0.51915 0.64688 0.55270 0.55591
## cg00000292 0.70312 0.67248 0.63095 0.64757 0.67921
## cg00000321 0.56316 0.54221 0.48866 0.63993 0.61028
## cg00000363 0.20862 0.24135 0.20399 0.22668 0.24959
## cg00000622 0.04034 0.03450 0.03127 0.03291 0.02887Phenotypes and/or covariates can be accessed using phenoData () function
#main variable used to be stored in:
table(pheno$characteristics_ch1)##
## tissue: prefrontal cortex tissue: superior temporal gyrus
## 142 144Note that phenoData() retrieves the subjects’ phenotypes in an AnnotatedDataFrame object which is converted to a data.frame by the function pData()
We can the, for instance, look at the CpGs located in the APP gene. To this end, the function fData () is used
## [1] "ID" "Name" "AddressA_ID" "AlleleA_ProbeSeq" "AddressB_ID"
## [6] "AlleleB_ProbeSeq" "Infinium_Design_Type" "Next_Base" "Color_Channel" "Forward_Sequence"
## [11] "Genome_Build" "CHR" "MAPINFO" "SourceSeq" "Chromosome_36"
## [16] "Coordinate_36" "Strand" "Probe_SNPs" "Probe_SNPs_10" "Random_Loci"
## [21] "Methyl27_Loci" "UCSC_RefGene_Name" "UCSC_RefGene_Accession" "UCSC_RefGene_Group" "UCSC_CpG_Islands_Name"
## [26] "Relation_to_UCSC_CpG_Island" "Phantom" "DMR" "Enhancer" "HMM_Island"
## [31] "Regulatory_Feature_Name" "Regulatory_Feature_Group" "DHS" "RANGE_START" "RANGE_END"
## [36] "RANGE_GB" "SPOT_ID"
#access to the names of genes in UCSC format
geneIDs <- annot$UCSC_RefGene_Name
genesIDs <- as.character(unlist(geneIDs))
selAPP <- grep("APP;APP",genesIDs)
length(selAPP)## [1] 21After that we can run different analyses. Also, several packages in Bioconductor have as input ExpressionSet objects.
1.2.2 SummarizedExperiment
The SummarizedExperiment class is a comprehensive data structure that can be used to store expression and methylation data from microarrays or read counts from RNA-seq experiments, among others. A SummarizedExperiment object contains slots for one or more datasets, feature annotation (e.g. genes, transcripts, SNPs, CpGs), individual phenotypes and experimental details, such as laboratory and experimental protocols. In a SummarizedExperiment, the rows of omic data are features and columns are subjects.

SummarizedExperiment
Information is coordinated across the object’s slots. For instance, subsetting samples in the assay matrix automatically subsets them in the phenotype metadata. A SummarizedExperimentobject is easily manipulated and constitutes the input and output of many of Bioconductor’s methods.
Data is retrieved from a SummarizedExperiment by using specific methods or accessors. We illustrate the functions with brge_methy which includes real methylation data and is available from the Bioconductor’s brgedata package. The data is made available by loading of the package
library(brgedata)
brge_methy## class: GenomicRatioSet
## dim: 392277 20
## metadata(0):
## assays(1): Beta
## rownames(392277): cg13869341 cg24669183 ... cg26251715 cg25640065
## rowData names(14): Forward_Sequence SourceSeq ... Regulatory_Feature_Group DHS
## colnames(20): x0017 x0043 ... x0077 x0079
## colData names(9): age sex ... Mono Neu
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: NA
## minfi version: NA
## Manifest version: NA
extends("GenomicRatioSet")## [1] "GenomicRatioSet" "RangedSummarizedExperiment" "SummarizedExperiment" "RectangularData" "Vector"
## [6] "Annotated" "vector_OR_Vector"The function extends() shows that the data has been encoded in an object of GenomicRatioSet class, which is an extension of the more primitive classes RangedSummarizedExperiment and SummarizedExperiment. brge_methy illustrates a typical object within Bioconductor’s framework, as it is a structure that inherits different types of classes in an established hierarchy. For each class, there are specific methods which are properly inherited across classes. For instance, in our example, SummarizedExperiment is brge_methy’s most primitive omic class and, therefore, all the methods of SummarizedExperiment apply to it. In particular, the methylation data that is stored in the object can be extracted with the function assay()
betas <- assay(brge_methy)
betas[1:5, 1:4]## x0017 x0043 x0036 x0041
## cg13869341 0.90331003 0.90082827 0.8543610 0.84231205
## cg24669183 0.86349463 0.84009629 0.8741255 0.85122181
## cg15560884 0.65986032 0.69634338 0.6931083 0.69752294
## cg01014490 0.01448404 0.01509478 0.0163614 0.01322362
## cg17505339 0.92176287 0.92060149 0.9101060 0.93249167The assay slot of a SummarizedExperiment object can contain any type of data (i.e. numeric, character, factor…), structure or large on-disk representations, such as a HDF5Array. Feature annotation data is accessed with the function rowData():
rowData(brge_methy)[,2:5]## DataFrame with 392277 rows and 4 columns
## SourceSeq Random_Loci Methyl27_Loci genes
## <character> <character> <character> <character>
## cg13869341 CCGGTGGCTGGCCACTCTGC.. WASH5P
## cg24669183 TCACCGCCTTGACAGCTTTG..
## cg15560884 CGCGTAAACAAGGGAAGCTG..
## cg01014490 TCAGAACTCGCGGTGGGGGC..
## cg17505339 AAACAAACAAAGATATCAAG..
## ... ... ... ... ...
## cg04964672 CGGCGGCTTTCCACGCTGCG..
## cg01086462 CGCAGGTATGGTGTACATTA..
## cg02233183 CAAAATGAATGAAATTTACA.. NLGN4Y;NLGN4Y
## cg26251715 CGCTGGTTGTCCAGGCTGGA.. TTTY14
## cg25640065 CGGGGCAGGGGTCCGTAACT..which returns a data.frame object. In our example, it contains the sequences and the genes associated with the CpG probes, among other information.
1.2.3 Genomic Ranges (GRanges)
The Bioconductor’s package GenomicRanges aims to represent and manipulate the genomic annotation of molecular omic data under a reference genome. It contains functions to select specific regions and perform operations with them (Lawrence 2013). Objects of GRanges class are important to annotate and manipulate genomic, transcriptomic and methylomic data. In particular, they are used in conjunction with SummarizedExperiment, within the RangedSummarizedExperiment class that is explained in the following section.
Annotation data refers to the characteristics of the variables in the high-dimensional data set. In particular for omic data relative to DNA structure and function, each variable may be given a location in a reference genome. While not two genomes are identical, the construction of a reference genome allows the mapping of specific characteristics of individual genomes to a common ground where they can be compared. The reference genome defines a coordinate system: “chromosome id” and “position along the chromosome”. For instance, a position such as chr10:4567-5671 would represent the 4567th to the 5671st base pair on the reference’s chromosome 10.
The main functionalities implemented GenomicRanges are methods on GRanges objects. Objects are created by the function GRanges, minimum requirements are the genomic positions given by the chromosome seqnames and base pair coordinates ranges. Other metadata (e.g. variables) can be added to provide further information about each segment.
We illustrate GenomicRanges creating 8 segments on either chr1 or chr2, each with defined start and end points. We add strand information, passed through the argument strand, to indicate the direction of each sequence. We also add a hypothetical variable disease that indicates whether asthma or obesity have been associated with each interval
library(GenomicRanges)
gr <- GRanges(seqnames=c(rep("chr1", 4), rep("chr2", 4)),
ranges = IRanges(start = c(1000, 1800, 5300, 7900,
1300, 2100, 3400, 6700),
end =c(2200, 3900, 5400, 8100,
2600, 3300, 4460, 6850)),
strand = rep(c("+", "-"), 4),
disease = c(rep("Asthma",4), rep("Obesity",4)))
gr## GRanges object with 8 ranges and 1 metadata column:
## seqnames ranges strand | disease
## <Rle> <IRanges> <Rle> | <character>
## [1] chr1 1000-2200 + | Asthma
## [2] chr1 1800-3900 - | Asthma
## [3] chr1 5300-5400 + | Asthma
## [4] chr1 7900-8100 - | Asthma
## [5] chr2 1300-2600 + | Obesity
## [6] chr2 2100-3300 - | Obesity
## [7] chr2 3400-4460 + | Obesity
## [8] chr2 6700-6850 - | Obesity
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsgr is our constructed object of class GRanges. The object responds to the usual array and subset extraction given by squared parentheses
gr[1]## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | disease
## <Rle> <IRanges> <Rle> | <character>
## [1] chr1 1000-2200 + | Asthma
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsHowever, there are also specific functions to access and modify information. For instance, seqnames() extract the chromosomes defined in our examples, whose first element can be redefined accordingly:
seqnames(gr)## factor-Rle of length 8 with 2 runs
## Lengths: 4 4
## Values : chr1 chr2
## Levels(2): chr1 chr2
seqnames(gr)[1] <- "chr2"
gr## GRanges object with 8 ranges and 1 metadata column:
## seqnames ranges strand | disease
## <Rle> <IRanges> <Rle> | <character>
## [1] chr2 1000-2200 + | Asthma
## [2] chr1 1800-3900 - | Asthma
## [3] chr1 5300-5400 + | Asthma
## [4] chr1 7900-8100 - | Asthma
## [5] chr2 1300-2600 + | Obesity
## [6] chr2 2100-3300 - | Obesity
## [7] chr2 3400-4460 + | Obesity
## [8] chr2 6700-6850 - | Obesity
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsThis is important to annotation using different system. NCBI encodes chromosomes as 1, 2, 3, …; while UCSC uses chr1, chr2, … The chromosome style can be changed using
seqlevelsStyle(gr) <- "NCBI"
gr## GRanges object with 8 ranges and 1 metadata column:
## seqnames ranges strand | disease
## <Rle> <IRanges> <Rle> | <character>
## [1] 2 1000-2200 + | Asthma
## [2] 1 1800-3900 - | Asthma
## [3] 1 5300-5400 + | Asthma
## [4] 1 7900-8100 - | Asthma
## [5] 2 1300-2600 + | Obesity
## [6] 2 2100-3300 - | Obesity
## [7] 2 3400-4460 + | Obesity
## [8] 2 6700-6850 - | Obesity
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsLet’s us write back the UCSC style
seqlevelsStyle(gr) <- "UCSC"
gr## GRanges object with 8 ranges and 1 metadata column:
## seqnames ranges strand | disease
## <Rle> <IRanges> <Rle> | <character>
## [1] chr2 1000-2200 + | Asthma
## [2] chr1 1800-3900 - | Asthma
## [3] chr1 5300-5400 + | Asthma
## [4] chr1 7900-8100 - | Asthma
## [5] chr2 1300-2600 + | Obesity
## [6] chr2 2100-3300 - | Obesity
## [7] chr2 3400-4460 + | Obesity
## [8] chr2 6700-6850 - | Obesity
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsAdditional information can be added to the current object as a new field of a list
gr$gene_id <- paste0("Gene", 1:8)
gr## GRanges object with 8 ranges and 2 metadata columns:
## seqnames ranges strand | disease gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr2 1000-2200 + | Asthma Gene1
## [2] chr1 1800-3900 - | Asthma Gene2
## [3] chr1 5300-5400 + | Asthma Gene3
## [4] chr1 7900-8100 - | Asthma Gene4
## [5] chr2 1300-2600 + | Obesity Gene5
## [6] chr2 2100-3300 - | Obesity Gene6
## [7] chr2 3400-4460 + | Obesity Gene7
## [8] chr2 6700-6850 - | Obesity Gene8
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsGenomicRanges provides different methods to perform arithmetic with the ranges, see ?GRanges for a full list. For instance, with shift() an interval is moved a given base-pair distance and with flank() the interval is stretched
#shift: move all intervals 10 base pair towards the end
GenomicRanges::shift(gr, 10)## GRanges object with 8 ranges and 2 metadata columns:
## seqnames ranges strand | disease gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr2 1010-2210 + | Asthma Gene1
## [2] chr1 1810-3910 - | Asthma Gene2
## [3] chr1 5310-5410 + | Asthma Gene3
## [4] chr1 7910-8110 - | Asthma Gene4
## [5] chr2 1310-2610 + | Obesity Gene5
## [6] chr2 2110-3310 - | Obesity Gene6
## [7] chr2 3410-4470 + | Obesity Gene7
## [8] chr2 6710-6860 - | Obesity Gene8
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
#shift: move each intervals individually
GenomicRanges::shift(gr, seq(10,100, length=8))## GRanges object with 8 ranges and 2 metadata columns:
## seqnames ranges strand | disease gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr2 1010-2210 + | Asthma Gene1
## [2] chr1 1822-3922 - | Asthma Gene2
## [3] chr1 5335-5435 + | Asthma Gene3
## [4] chr1 7948-8148 - | Asthma Gene4
## [5] chr2 1361-2661 + | Obesity Gene5
## [6] chr2 2174-3374 - | Obesity Gene6
## [7] chr2 3487-4547 + | Obesity Gene7
## [8] chr2 6800-6950 - | Obesity Gene8
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
#flank: recover regions next to the input set.
# For a 50 base stretch upstream (negative value for
# downstream)
flank(gr, 50)## GRanges object with 8 ranges and 2 metadata columns:
## seqnames ranges strand | disease gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr2 950-999 + | Asthma Gene1
## [2] chr1 3901-3950 - | Asthma Gene2
## [3] chr1 5250-5299 + | Asthma Gene3
## [4] chr1 8101-8150 - | Asthma Gene4
## [5] chr2 1250-1299 + | Obesity Gene5
## [6] chr2 3301-3350 - | Obesity Gene6
## [7] chr2 3350-3399 + | Obesity Gene7
## [8] chr2 6851-6900 - | Obesity Gene8
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsGenomicRanges also includes methods for aggregating and summarizing GRangesobjects. The functions reduce(), disjoint() and coverage() are most useful. disjoin(), for instance, reduces the intervals into the smallest set of unique, non-overlapping pieces that make up the original object. It is strand-specific by default, but this can be avoided with ignore.strand=TRUE.
disjoin(gr, ignore.strand=TRUE)## GRanges object with 10 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 1800-3900 *
## [2] chr1 5300-5400 *
## [3] chr1 7900-8100 *
## [4] chr2 1000-1299 *
## [5] chr2 1300-2099 *
## [6] chr2 2100-2200 *
## [7] chr2 2201-2600 *
## [8] chr2 2601-3300 *
## [9] chr2 3400-4460 *
## [10] chr2 6700-6850 *
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsreduce() creates the smallest range set of unique, non-overlapping intervals. Strand information is also taken into account by default and can also be turned off
GenomicRanges::reduce(gr, ignore.strand=TRUE)## GRanges object with 6 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 1800-3900 *
## [2] chr1 5300-5400 *
## [3] chr1 7900-8100 *
## [4] chr2 1000-3300 *
## [5] chr2 3400-4460 *
## [6] chr2 6700-6850 *
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthscoverage() summarizes the times each base is covered by an interval
coverage(gr)## RleList of length 2
## $chr1
## integer-Rle of length 8100 with 6 runs
## Lengths: 1799 2101 1399 101 2499 201
## Values : 0 1 0 1 0 1
##
## $chr2
## integer-Rle of length 6850 with 10 runs
## Lengths: 999 300 800 101 400 700 99 1061 2239 151
## Values : 0 1 2 3 2 1 0 1 0 1It is also possible to perform operations between two different GRanges objects. For instance, one may be interested in knowing the intervals that overlap with a targeted region:
## GRanges object with 1 range and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 1200-4000 *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
gr.ov <- findOverlaps(target, gr)
gr.ov## Hits object with 1 hit and 0 metadata columns:
## queryHits subjectHits
## <integer> <integer>
## [1] 1 2
## -------
## queryLength: 1 / subjectLength: 8To recover the overlapping intervals between gr and target we can run
gr[subjectHits(gr.ov)]## GRanges object with 1 range and 2 metadata columns:
## seqnames ranges strand | disease gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr1 1800-3900 - | Asthma Gene2
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsor
subsetByOverlaps(gr, target)## GRanges object with 1 range and 2 metadata columns:
## seqnames ranges strand | disease gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr1 1800-3900 - | Asthma Gene2
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsOther operations can be found here.
1.2.4 RangedSummarizedExperiment
SummarizedExperiment is extended to RangedSummarizedExperiment, a child class that contains the annotation data of the features in a GenomicRanges object. In our epigenomic example, the second most primitive class of brge_methy object with omic functionality, after SummarizedExperiment, is RangedSummarizedExperiment. Annotation data, with variable names given by
names(rowData(brge_methy))## [1] "Forward_Sequence" "SourceSeq" "Random_Loci" "Methyl27_Loci" "genes"
## [6] "UCSC_RefGene_Accession" "group" "Phantom" "DMR" "Enhancer"
## [11] "HMM_Island" "Regulatory_Feature_Name" "Regulatory_Feature_Group" "DHS"can be obtained in a GRanges object, for a given variable. For instance, metadata of CpG genomic annotation and neighboring genes is obtained using array syntax
rowRanges(brge_methy)[, "genes"]## GRanges object with 392277 ranges and 1 metadata column:
## seqnames ranges strand | genes
## <Rle> <IRanges> <Rle> | <character>
## cg13869341 chr1 15865 * | WASH5P
## cg24669183 chr1 534242 * |
## cg15560884 chr1 710097 * |
## cg01014490 chr1 714177 * |
## cg17505339 chr1 720865 * |
## ... ... ... ... . ...
## cg04964672 chrY 7428198 * |
## cg01086462 chrY 7429349 * |
## cg02233183 chrY 16634382 * | NLGN4Y;NLGN4Y
## cg26251715 chrY 21236229 * | TTTY14
## cg25640065 chrY 23569324 * |
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengthsSubject data can be accessed entirely in a single data.frame or a variable at the time. The entire subject (phenotype) information is retrieved with the function colData():
colData(brge_methy)## DataFrame with 20 rows and 9 columns
## age sex NK Bcell CD4T CD8T Eos Mono Neu
## <numeric> <factor> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## x0017 4 Female -5.81386e-19 0.1735078 0.20406869 0.1009741 0.00000e+00 0.0385654 0.490936
## x0043 4 Female 1.64826e-03 0.1820172 0.16171272 0.1287722 0.00000e+00 0.0499542 0.491822
## x0036 4 Male 1.13186e-02 0.1690173 0.15546368 0.1277417 0.00000e+00 0.1027105 0.459632
## x0041 4 Female 8.50822e-03 0.0697158 0.00732789 0.0321861 0.00000e+00 0.0718280 0.807749
## x0032 4 Male 0.00000e+00 0.1139780 0.22230399 0.0216090 2.60504e-18 0.0567246 0.575614
## ... ... ... ... ... ... ... ... ... ...
## x0018 4 Female 0.0170284 0.0781698 0.112735 0.06796816 8.37770e-03 0.0579535 0.658600
## x0057 4 Female 0.0000000 0.0797774 0.111072 0.00910489 4.61887e-18 0.1015260 0.686010
## x0061 4 Female 0.0000000 0.1640266 0.224203 0.13125212 8.51074e-03 0.0382647 0.429575
## x0077 4 Female 0.0000000 0.1122731 0.168056 0.07840593 6.13397e-02 0.0583411 0.515284
## x0079 4 Female 0.0120148 0.0913650 0.205830 0.11475389 0.00000e+00 0.0750535 0.513597The list symbol $ can be used, for instance, to obtain the sex of the individuals
brge_methy$sex## [1] Female Female Male Female Male Male Male Male Male Male Female Female Female Male Male Female Female Female Female Female
## Levels: Female MaleSubsetting the entire structure is also possible following the usual array syntax. For example, we can select only males from the brge_methy dataset
brge_methy[, brge_methy$sex == "male"]## class: GenomicRatioSet
## dim: 392277 0
## metadata(0):
## assays(1): Beta
## rownames(392277): cg13869341 cg24669183 ... cg26251715 cg25640065
## rowData names(14): Forward_Sequence SourceSeq ... Regulatory_Feature_Group DHS
## colnames(0):
## colData names(9): age sex ... Mono Neu
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: NA
## minfi version: NA
## Manifest version: NAThe metadata() function retrieves experimental data
metadata(brge_methy)## list()which in our case is empty.